library(Seurat)# 读取PBMC数据集counts<-Read10X(data.dir ="data/seurat_official/filtered_gene_bc_matrices/hg19")# Initialize the Seurat object with the raw (non-normalized data).pbmc<-CreateSeuratObject(counts =counts, project ="pbmc3k", min.cells =3, min.features =200)pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
An object of class Seurat
31130 features across 29629 samples within 3 assays
Active assay: integrated (3000 features, 3000 variable features)
2 layers present: data, scale.data
2 other assays present: RNA, SCT
2 dimensional reductions calculated: pca, umap
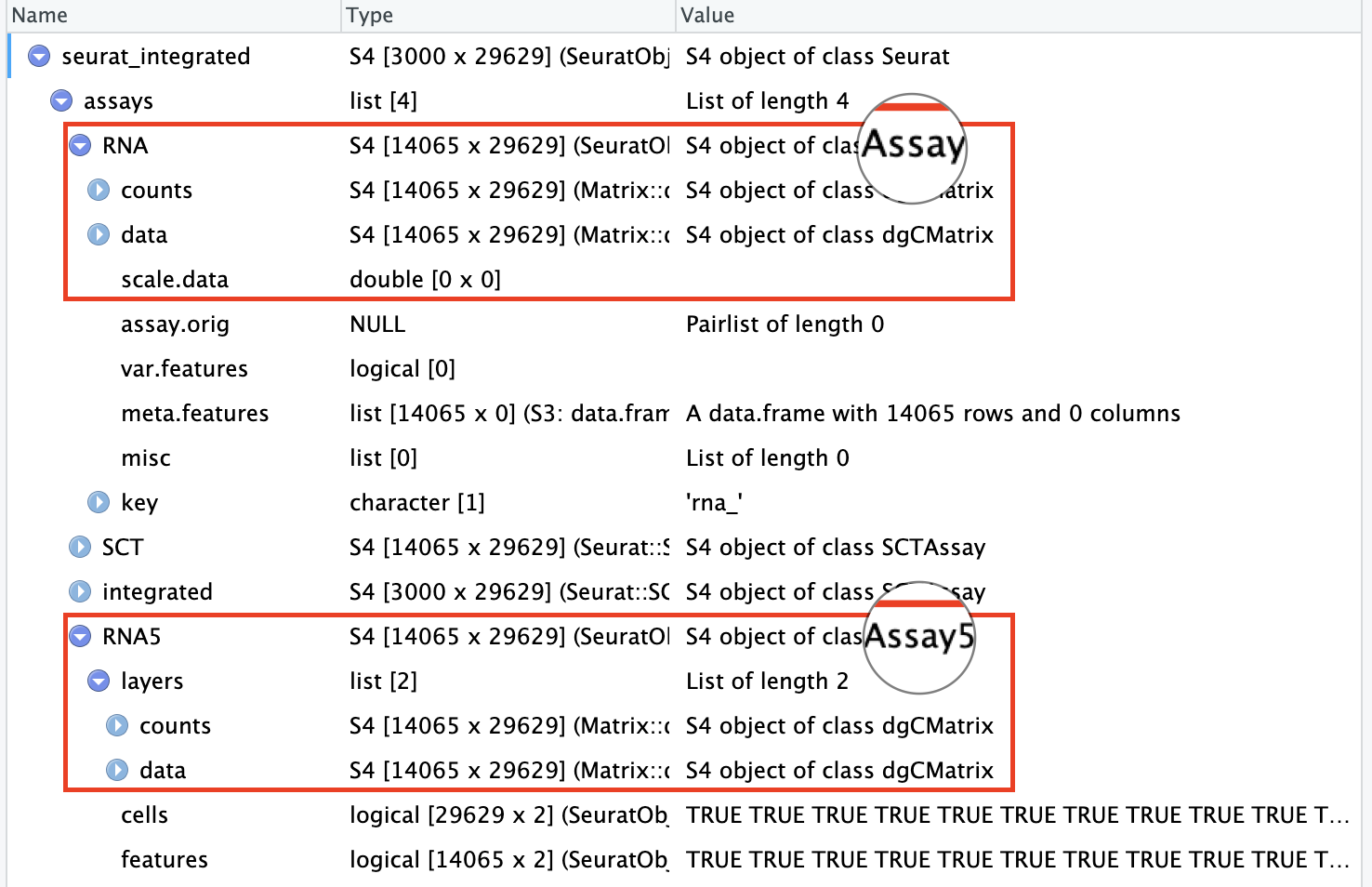
# convert a v4 or v3 assay to a v5 assayseurat_integrated[["RNA5"]]<-as(object =seurat_integrated[["RNA"]], Class ="Assay5")DefaultAssay(seurat_integrated)="RNA5"
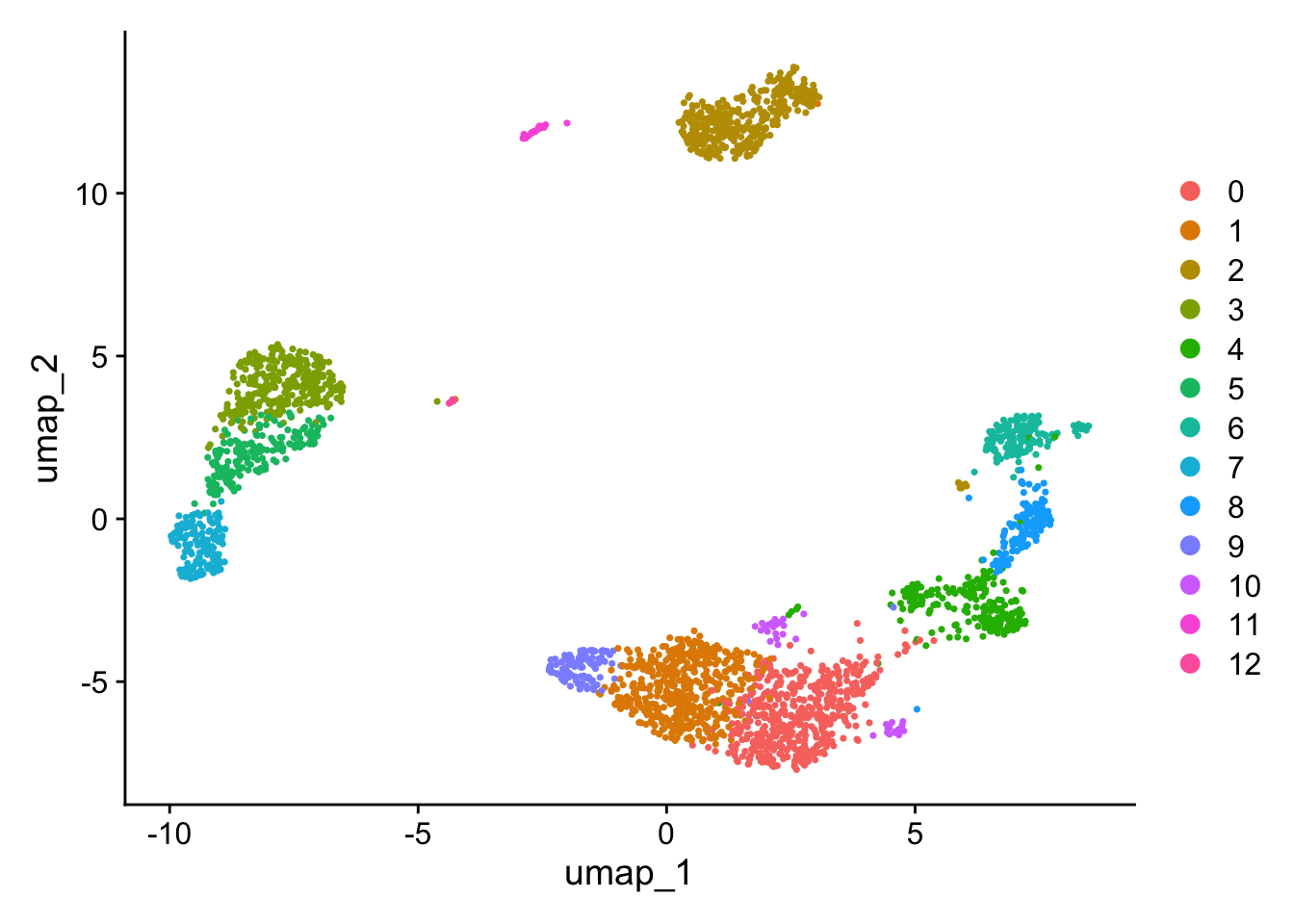
[1] CD16 Mono DC pDC B Activated B
[6] CD8 T Mk CD14 Mono T activated CD4 Naive T
[11] CD4 Memory T NK epi
13 Levels: pDC Mk DC CD14 Mono CD16 Mono B Activated B CD8 T NK ... epi
[1] CD16 Mono DC pDC B Activated B cell
[6] CD8 T Mk CD14 Mono T activated CD4 Naive T
[11] CD4 Memory T NK epi
13 Levels: B cell pDC Mk DC CD14 Mono CD16 Mono B Activated CD8 T ... epi
2.3 获取meta.data
# View metadata data frame, stored in object@meta.datapbmc@meta.data|>head()
# Set expression data assume new.data is a new expression matrixpbmc[["RNA"]]$counts<-new.data# 或LayerData(pbmc, assay ="RNA", layer ="counts")<-new.data
FetchData can access anything from expression matrices, cell embeddings, or metadata use the previously listed commands to access entire matrices。通过FetchData可以提取包括表达量数据、PCA分数以及meta.data内的任何变量并形成一个数据框。实际应用场景见分析主成分(PCs)对细胞分群的影响。
FetchData(object =pbmc, vars =c("PC_1", "nFeature_RNA", "MS4A1"), layer ="counts")|>head()
An object of class Seurat
40000 features across 1350 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
# 提取特定cell identities, also see ?SubsetDatasubset(x =pbmc, idents ="B cell")
An object of class Seurat
40000 features across 157 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
An object of class Seurat
40000 features across 2505 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
An object of class Seurat
40000 features across 519 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
An object of class Seurat
40000 features across 517 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
An object of class Seurat
40000 features across 45 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
# Downsample the number of cells per identity classsubset(x =pbmc, downsample =100)
An object of class Seurat
40000 features across 1088 samples within 3 assays
Active assay: SCT (12572 features, 3000 variable features)
3 layers present: counts, data, scale.data
2 other assays present: RNA, RNA3
2 dimensional reductions calculated: pca, umap
3.2 分割layers
In Seurat v5, users can now split in object directly into different layers keeps expression data in one object, but splits multiple samples into layers can proceed directly to integration workflow after splitting layers。实际应用场景见数据整合。
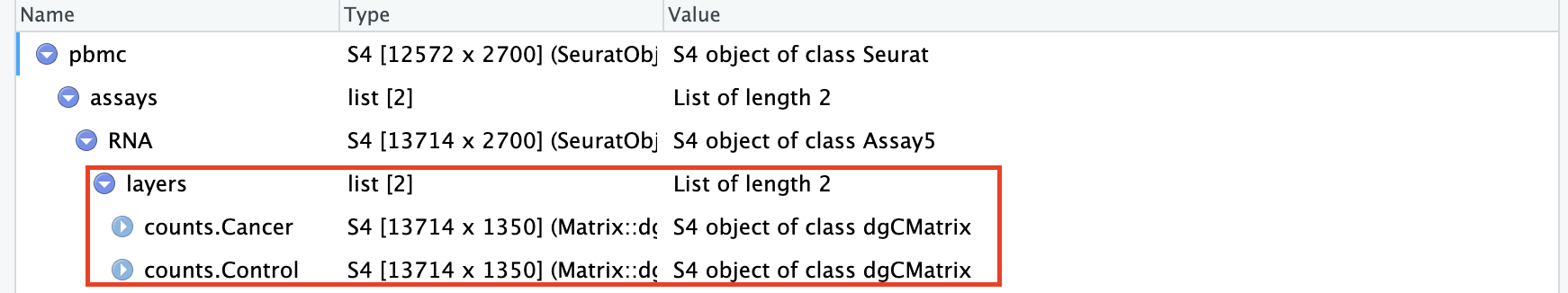
DefaultAssay(pbmc)<-"RNA"# 按照meta.data中的stim列分割layerpbmc[["RNA"]]<-split(pbmc[["RNA"]], f =pbmc$groups)
In line with prior workflows, you can also split your Seurat object into a list of multiple objects based on a metadata column creates a list of two objects。通过SplitObject()分割Seurat之后生成的是包含多个Seurat对象的列表。
$Cancer
An object of class Seurat
40000 features across 1350 samples within 3 assays
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
2 other assays present: SCT, RNA3
2 dimensional reductions calculated: pca, umap
$Control
An object of class Seurat
40000 features across 1350 samples within 3 assays
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
2 other assays present: SCT, RNA3
2 dimensional reductions calculated: pca, umap
3.4 Merge objects (without integration)
In Seurat v5, merging creates a single object, but keeps the expression information split into different layers for integration. If not proceeding with integration, rejoin the layers after merging. 实际应用场景,见后续章节。
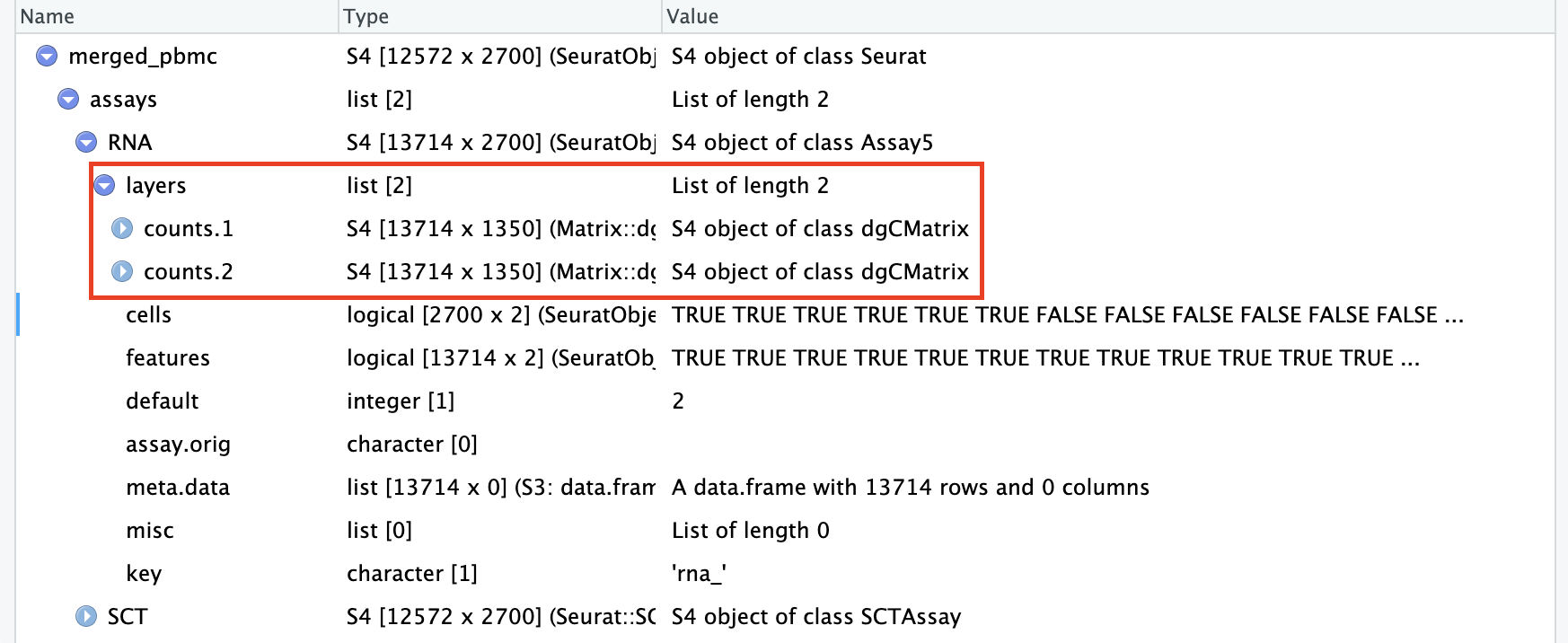
# Merge two Seurat objectsmerged_pbmc<-merge(x =seurat_list[["Control"]], y =seurat_list[["Cancer"]])
# Example to merge more than two Seurat objectsmerge(x =pbmc1, y =list(pbmc2, pbmc3))
merged_pbmc<-NormalizeData(merged_pbmc, verbose =F)merged_pbmc<-FindVariableFeatures(merged_pbmc, verbose =F)merged_pbmc<-ScaleData(merged_pbmc, verbose =F)merged_pbmc<-RunPCA(merged_pbmc, verbose =F)merged_pbmc<-IntegrateLayers(object =merged_pbmc, method =RPCAIntegration, orig.reduction ="pca", new.reduction ="integrated.rpca", verbose =FALSE)# now that integration is complete, rejoin layersmerged_pbmc[["RNA"]]<-JoinLayers(merged_pbmc[["RNA"]])merged_pbmc
An object of class Seurat
40000 features across 2700 samples within 3 assays
Active assay: RNA (13714 features, 2000 variable features)
3 layers present: data, counts, scale.data
2 other assays present: SCT, RNA3
2 dimensional reductions calculated: pca, integrated.rpca
Additional resources
Users who are particularly interested in some of the technical changes to data storage in Seurat v5 can explore the following resources: